
摘要
本发明提供了一种吖啶二酮类化合物在制备抗糖尿病药物中的应用,属于生物医药技术领域。本发明首次证实了吖啶二酮类化合物可以通过激活并上调GPR40蛋白表达,参与GPR40‑PPARγ‑PI3K/Akt‑GLUT4信号通路,促进胰岛素分泌,增加肝脏和肌肉组织的葡萄糖消耗,改善胰岛素抵抗。吖啶二酮类化合物的作用靶点为GPR40受体,其促胰岛素分泌作用具有葡萄糖依赖性,当外周血糖低于一定程度时,其降糖作用会消失。将吖啶二酮类化合物制备成抗糖尿药物,将为糖尿病的治疗提供全新选择和策略。
权利要求书
1.一种吖啶二酮类化合物或其药学可接受的盐或其药学上可接受的酯,其特征在于,该化合物的结构如下:每个R1和R2独立为氢、C1~C3烷基、-COOH、-CH2Ph,-CH2CH(CH3)2,-Ph,或X=O,N或S;每个R3独立为氢、卤素、-CF3、C1~C4烷基或烷氧基、-NO2或-OH;每个R4独立为氢、卤素、-CF3、C1~C3烷基或烷氧基、-NO2或-OH;R5为-COOH,-COOCH3或-COOC2H5;n为0、1或2。
2.一种吖啶二酮类化合物或其药学可接受的盐或其药学上可接受的酯在制备抗糖尿病药物中的应用,其中,所述吖啶二酮类化合物的结构如下所示;每个R1和R2独立为氢、C1~C3烷基、-COOH、-CH2Ph,-CH2CH(CH3)2,-Ph,或X=O,N或S;每个R3独立为氢、卤素、-CF3、C1~C4烷基或烷氧基、-NO2或-OH;每个R4独立为氢、卤素、-CF3、C1~C3烷基或烷氧基、-NO2或-OH;R5为-COOH,-COOCH3或-COOC2H5;n为0、1或2。
3.根据权利要求2所述的一种吖啶二酮类化合物或其药学可接受的盐或其药学上可接受的酯在制备抗糖尿病药物中的应用,其特征在于,抗糖尿病药物为GPR40激动剂。
4.根据权利要求2所述的一种吖啶二酮类化合物或其药学可接受的盐或其药学上可接受的酯在制备抗糖尿病药物中的应用,其特征在于,抗糖尿病药物为葡萄糖依赖性促胰岛素分泌药物。
5.根据权利要求2所述的一种吖啶二酮类化合物或其药学可接受的盐或其药学上可接受的酯在制备抗糖尿病药物中的应用,其特征在于,抗糖尿病药物为临床可接受的药物制剂。
6.根据权利要求5所述的一种吖啶二酮类化合物或其药学可接受的盐或其药学上可接受的酯在制备抗糖尿病药物中的应用,其特征在于,药物制剂为片剂、胶囊剂、颗粒剂或注射剂。
7.一种吖啶二酮类化合物或其药学可接受的盐或其药学上可接受的酯在制备GPR40激动剂中的应用,其中,该化合物的结构如下:每个R1和R2独立为氢、C1~C3烷基、-COOH、-CH2Ph,-CH2CH(CH3)2,-Ph,或X=O,N或S;每个R3独立为氢、卤素、-CF3、C1~C4烷基或烷氧基、-NO2或-OH;每个R4独立为氢、卤素、-CF3、C1~C3烷基或烷氧基、-NO2或-OH;R5为-COOH,-COOCH3或-COOC2H5;n为0、1或2。
[收起]
说明书
技术领域
本发明属于生物医药技术领域,特别涉及吖啶二酮类化合物在制备抗糖尿病药物中的应用。
背景技术
目前全球20至79岁的成年人中约有4.63亿人患有糖尿病,其中2型糖尿病(T2DM)患者的数量占糖尿病患者总数的90%以上。据统计2019年因糖尿病及其并发症导致的死亡人数高达420万。中国糖尿病患者已达到1.16亿,约占全球糖尿病人总数的四分之一,2019年我国因糖尿病及其并发症导致的死亡人数约为82.3万。2型糖尿病已经成为影响人类健康的重要问题。传统抗2型糖尿病药物发挥降糖作用主要通过刺激胰岛β细胞分泌胰岛素以及提高胰岛素敏感性,这些作用与外周血糖无关,因此使用这些药物降血糖的同时会增加患者发生低血糖的风险。GPR40是G蛋白偶联受体家族成员之一,主要分布于胰岛β细胞、肠道K和L细胞,是脂肪酸特异性受体。GPR40介导的胰岛素分泌具有葡萄糖依赖性,当外周血糖低于一定程度时,其降糖作用会消失,从而减少低血糖发生率。GPR40受体因其独特的血糖调节作用,已成为抗2型糖尿病药物开发的潜在靶点,开发利用GPR40激动剂或先导化合物对抗2型糖尿病药物的开发具有重要意义。
发明内容
本发明的目的在于提供一种吖啶二酮类化合物在制备抗糖尿病药物中的应用,为糖尿病治疗提供了全新的选择和策略。
本发明通过以下技术方案来实现:
一种吖啶二酮类化合物或其药学可接受的盐或其药学上可接受的酯,该化合物的结构如下:
每个R1和R2独立为氢、C1~C3烷基、-COOH、-CH2Ph,-CH2CH(CH3)2,-Ph,或X=O,N或S;
每个R3独立为氢、卤素、-CF3、C1~C4烷基或烷氧基、-NO2或-OH;
每个R4独立为氢、卤素、-CF3、C1~C3烷基或烷氧基、-NO2或-OH;
R5为-COOH,-COOCH3或-COOC2H5;
n为0、1或2。
一种吖啶二酮类化合物或其药学可接受的盐或其药学上可接受的酯在制备抗糖尿病药物中的应用,其中,所述吖啶二酮类化合物的结构如下所示;
每个R1和R2独立为氢、C1~C3烷基、-COOH、-CH2Ph,-CH2CH(CH3)2,-Ph,或X=O,N或S;
每个R3独立为氢、卤素、-CF3、C1~C4烷基或烷氧基、-NO2或-OH;
每个R4独立为氢、卤素、-CF3、C1~C3烷基或烷氧基、-NO2或-OH;
R5为-COOH,-COOCH3或-COOC2H5;
n为0、1或2。
本发明进一步的改进在于,抗糖尿病药物为GPR40激动剂。
本发明进一步的改进在于,抗糖尿病药物为葡萄糖依赖性促胰岛素分泌药物。
本发明进一步的改进在于,抗糖尿病药物为临床可接受的药物制剂。
本发明进一步的改进在于,药物制剂为片剂、胶囊剂、颗粒剂或注射剂。
一种吖啶二酮类化合物或其药学可接受的盐或其药学上可接受的酯在制备GPR40激动剂中的应用,其中,该化合物的结构如下:
每个R1和R2独立为氢、C1~C3烷基、-COOH、-CH2Ph,-CH2CH(CH3)2,-Ph,或X=O,N或S;
每个R3独立为氢、卤素、-CF3、C1~C4烷基或烷氧基、-NO2或-OH;
每个R4独立为氢、卤素、-CF3、C1~C3烷基或烷氧基、-NO2或-OH;
R5为-COOH,-COOCH3或-COOC2H5;
n为0、1或2。
与现有技术相比,本发明具有以下有益的技术效果:
本发明首次发现吖啶二酮类化合物或其药学可接受的盐能够在制备抗糖尿病药物中的应用,并证实了吖啶二酮类化合物可以通过激活并上调GPR40蛋白表达,参与GPR40-PPARγ-PI3K/Akt-GLUT4信号通路,促进胰岛素分泌,增加肝脏和肌肉组织的葡萄糖消耗,改善胰岛素抵抗,发挥抗2型糖尿病的作用。吖啶二酮类化合物的作用靶点为GPR40受体,其促胰岛素分泌作用具有葡萄糖依赖性,当外周血糖低于一定程度时,其降糖作用会消失。将吖啶二酮类化合物制备成抗糖尿药物,将为糖尿病的治疗提供全新选择和策略。
附图说明
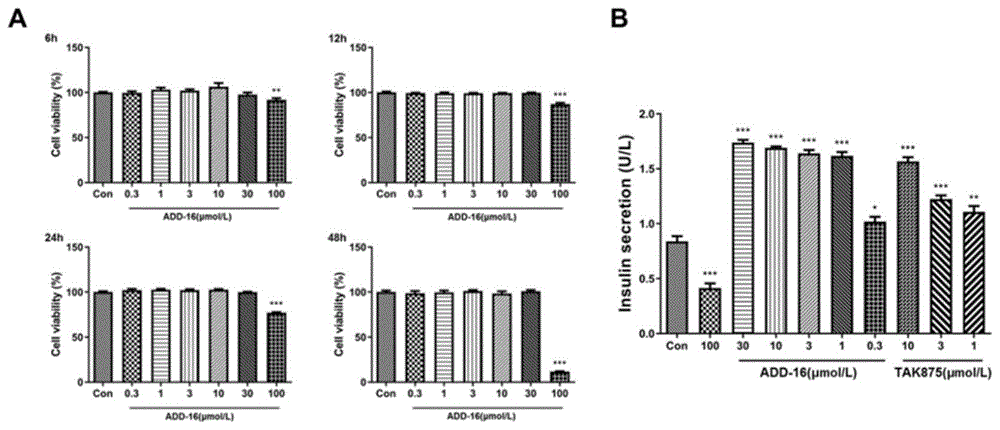
图1为ADD-16促进MIN6细胞中葡萄糖刺激的胰岛素分泌。其中,A为细胞毒性,包括6h、12h、24h与48h;B为胰岛素分泌。±s(n=6),*P<0.05,**P<0.01,***P<0.001vs.Con组。
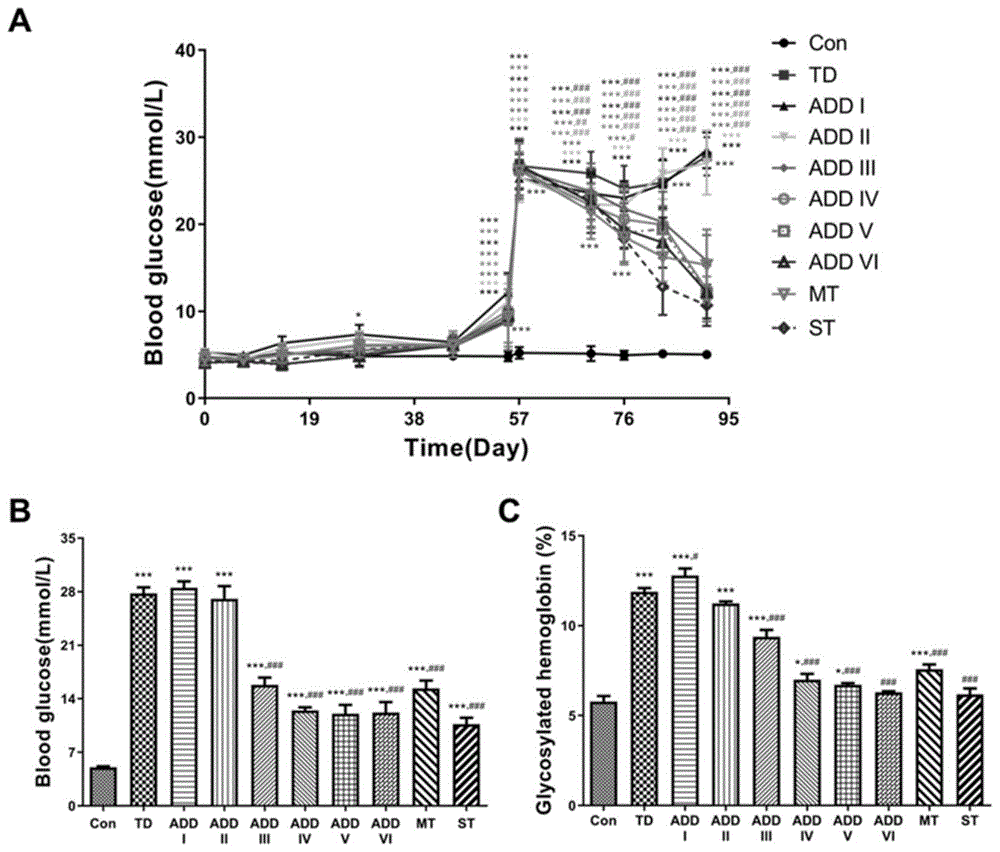
图2为ADD-16对STZ诱导T2DM大鼠血糖调节的影响。其中,A为餐后血糖变化趋势;B为实验结束时各组大鼠餐后血糖值;C为糖化血红蛋白值。±s(n=10),*P<0.05,**P<0.01,***P<0.001vs.Con组;#P<0.05,##P<0.01,###P<0.001vs.TD组。
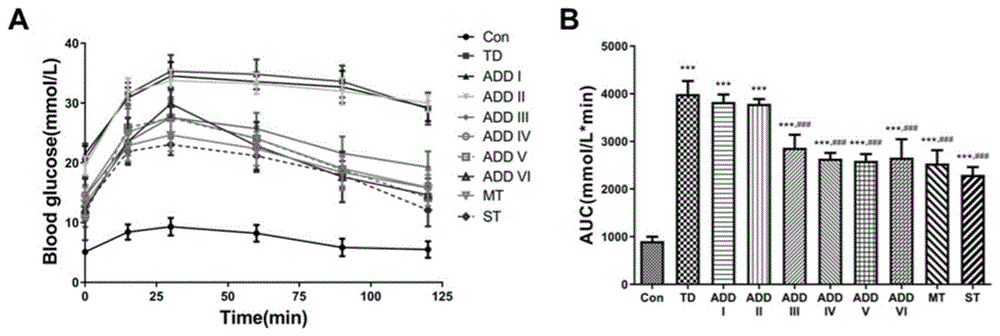
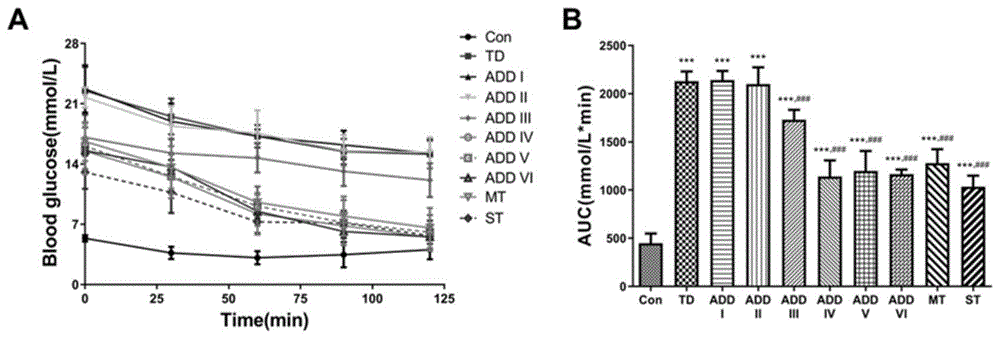
图3为ADD-16对STZ诱导T2DM大鼠糖耐量的影响。其中,A为OGTT曲线;B为曲线下面积。±s(n=10),*P<0.05,**P<0.01,***P<0.001vs.Con组;#P<0.05,##P<0.01,###P<0.001vs.TD组。
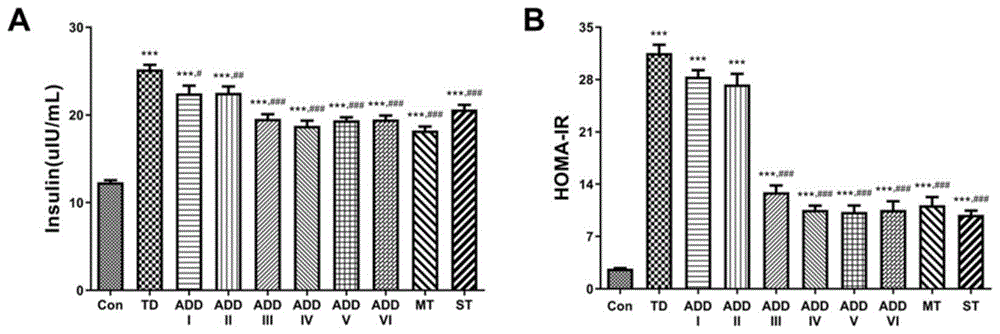
图4为ADD-16改善STZ诱导T2DM大鼠胰岛素抵抗。其中,A为血清胰岛素含量;B为胰岛素抵抗指数。±s(n=10),*P<0.05,**P<0.01,***P<0.001vs.Con组;#P<0.05,##P<0.01,###P<0.001vs.TD组。
图5为ADD-16对STZ诱导T2DM大鼠胰岛素耐量的影响。其中,A为ITT曲线;B为曲线下面积。±s(n=10),*P<0.05,**P<0.01,***P<0.001vs.Con组;#P<0.05,#P<0.01,###P<0.001vs.TD组。
图6为ADD-16对STZ诱导T2DM大鼠脂代谢的影响。其中,A为FFA;B为TG;C为TC;D为LDL;E为HDL的含量。±s(n=10),*P<0.05,**P<0.01,***P<0.001vs.Con组;#P<0.05,##P<0.01,###P<0.001vs.TD组。
图7为大鼠单次口服ADD-16后血药浓度-时间曲线(±s,n=8)。
图8为大鼠单次口服ADD-16后各时间点组织浓度分布图(±s,n=6)。
图9为ADD-16通过GPR40改善MIN6细胞胰岛素抵抗。其中,A为分组处理24h后各组胰岛素分泌;B为不同给药条件下胰岛素分泌水平的变化。±s(n=6),*P<0.05,**P<0.01,***P<0.001vs.Con组;#P<0.05,##P<0.01,###P<0.001vs.IR组。
图10为ADD-16对ZDF大鼠胰岛组织中胰岛素信号相关分子表达的影响。±s(n=3),*P<0.05,**P<0.01,***P<0.001vs.Con组;#P<0.05,##P<0.01,###P<0.001vs.TD组。
图11为ADD-16对MIN6中胰岛素信号相关分子表达的影响。其中,A为MIN6细胞中的WB结果;B为MIN6细胞中GPR40的免疫荧光染色结果。±s(n=3),*P<0.05,**P<0.01,***P<0.001vs.Con组;#P<0.05,##P<0.01,###P<0.001vs.IR组。
图12为32个化合物结合GPR40构象叠合模式。
具体实施方式
下面结合具体的实施例对发明做进一步的详细说明,所述是对本发明的解释而不是限定。
一种吖啶二酮类化合物或其药学可接受的盐或其药学上可接受的酯在制备抗糖尿病药物中的应用,其中,所述吖啶二酮类化合物的结构如下所示。
每个R1和R2独立为氢、C1-3烷基、COOH、CH2Ph,CH2CH(CH3)2,Ph,或(X=O,N,S);
每个R3独立为氢、卤素、CF3、C1-4烷基或烷氧基、NO2或OH;
每个R4独立为氢、卤素、CF3、C1-4烷基或烷氧基、NO2或OH;
R5为羧基、羧酸钾、羧酸钠或羧酸钙、羧酸甲酯或乙酯;
n为0、1或2
优选地,所述抗糖尿病药物为GPR40激动剂。
优选地,所述抗糖尿病药物为葡萄糖依赖性促胰岛素分泌药物。
优选地,所述抗糖尿病药物为临床可接受的药物制剂。
优选地,所述药物制剂为片剂、胶囊剂、颗粒剂等其他口服剂型或注射剂型。
一种吖啶二酮类化合物或其药学可接受的盐或其药学上可接受的酯在制备GPR40激动剂中的应用。
吖啶二酮类化合物的药学可接受的盐为羧酸钾、羧酸钠或羧酸钙,即R5为羧酸钾、羧酸钠或羧酸钙、羧酸甲酯或羧酸乙酯。
1.吖啶二酮类GPR40激动剂的合成
以ADD-16、ADD-17、ADD-18与ADD-19的合成为例,合成路线如下:
以ADD-16合成为例。
将5,5-二甲基-1,3-环已二酮(2.8g,20mmol)、3-溴-4-羟基苯甲醛(2.01g,10mmol)和乙酸铵(2.31g,30mmol)置于100mL容量瓶中,再加入未经纯化的离子液体催化剂(0.2g,0.36mmol),80℃回流4h,TLC监测反应,反应结束后降至室温,过滤,无水乙醇冲洗,干燥,得吖啶二酮中间体粗产物3.79g,产率约83.3%,不纯化,直接用于下一步反应。
50mL DMSO加入圆底烧瓶中,红外灯下将KOH(3.06g,0.055mol)研为粉末,加入并搅拌10min,加入制备的吖啶二酮中间体(3.79g,9.1mmol),搅拌15min,再加入对溴甲基苯甲酸(1.96g,9.1mmol),室温反应4h,TLC监测反应,反应结束后加入蒸馏水50mL,浓盐酸调pH至5.0,EtOAc萃取,合并有机层,无水Na2SO4干燥过夜,快速柱层析纯化(石油醚:乙酸乙酯/1:9V:V),得淡黄色固体2.74g,产率约52.1%。
ADD-16(R1=R2=CH3,m-R3=Br,R4=H,R5=COOH,n=1),核磁数据1H NMR(400MHz,DMSO-d6)δ9.323(s,1H),7.963(d,J=8.4Hz,2H),7.559(d,J=8.4Hz,2H),7.336(d,J=2.0Hz,1H),7.088(dd,J=2.0,2.0Hz,1H),7.007(dd,J=8.3,8.8Hz,1H),4.67(s,1H),2.453(d,J=16.8Hz,2H),2.345(d,J=16.8Hz,2H),2.181(d,J=16.0Hz,2H),2.001(d,J=16.0Hz,3H),1.014(s,6H),0.8886(s,6H);13C NMR(126MHz,DMSO-d6)δ194.89,167.53,152.60,149.89,142.26,142.00,132.54,130.72,129.92、128.29、113.93、111.58、110.68、79.94,69.90、50.64、32.63、29.52、26.92。高分辨质谱数据HRMS(ESI+)for C31H32BrNO5[M+H]+:m/z cale.,578.1537;found,578.1529。
以相同的方法合成得到ADD-17(R1=R2=CH3,m-R3=Cl,R4=H,R5=COOH,n=1),核磁数据1H NMR(400MHz,DMSO-d6),核磁数据:1H NMR(400MHz,DMSO-d6)δ9.292(s,1H),7.963(d,J=8.3Hz,2H),7.559(d,J=8.5Hz,2H),7.183(d,J=2.1Hz,1H),6.988(dd,J=2.0,2.0Hz,1H),6.847(dd,J=8.3,8.8Hz,1H),4.69(s,1H),2.443(d,J=17.8Hz,2H),2.315(d,J=17.0Hz,2H),2.171(d,J=16.1Hz,2H),1.991(d,J=16.2Hz,2H),1.004(s,6H),0.8786(s,6H);13C NMR(126MHz,DMSO-d6)δ194.49,167.03,152.10,149.49,141.86,141.50,132.04,130.72,129.92、128.29、113.43、111.08、110.08、79.94,69.90、52.64、32.13、29.02、26.52。高分辨质谱数据HRMS(ESI+)for C31H32ClNO5[M+H]+:m/z cale.,533.1969;found,533.1959。
以相同的方法合成得到ADD-18(R1=R2=CH3,m-R3=CF3,R4=H,R5=COOH,n=1),核磁数据:1H NMR(400MHz,DMSO-d6)δ9.523(s,1H),8.163(d,J=8.2Hz,2H),8.129(d,J=8.0Hz,2H),7.666(d,J=2.2Hz,1H),7.498(dd,J=2.1,2.0Hz,1H),7.157(dd,J=8.5,8.9Hz,1H),4.79(s,1H),2.433(d,J=16.5Hz,2H),2.345(d,J=16.6Hz,2H),2.081(d,J=16.1Hz,2H),2.011(d,J=16.0Hz,2H),1.024(s,6H),0.8986(s,6H);13C NMR(126MHz,DMSO-d6)δ194.19,169.53,153.60,150.89,144.26,143.00,134.54,132.72,130.92、129.29、114.93、112.58、111.68、80.94,70.90、50.84、32.83、29.72、27.32。高分辨质谱数据HRMS(ESI+)for C32H32F3NO5[M+H]+:m/z cale.,567.2233;found,567.2229。
以相同的方法合成得到ADD-19(R1=R2=CH3,o-R3=Br,R4=H,R5=COOH,n=1),核磁数据:1H NMR(400MHz,DMSO-d6)δ9.223(s,1H),8.143(d,J=8.2Hz,2H),8.129(d,J=8.0Hz,2H),7.496(d,J=2.2Hz,1H),7.338(dd,J=2.1,2.0Hz,1H),7.307(dd,J=8.5,8.9Hz,1H),4.78(s,1H),2.433(d,J=16.5Hz,2H),2.345(d,J=16.6Hz,2H),2.081(d,J=16.1Hz,2H),2.011(d,J=16.0Hz,2H),1.024(s,6H),0.8986(s,6H);13C NMR(126MHz,DMSO-d6)δ195.19,168.53,154.60,151.89,143.26,142.00,133.54,131.72,130.92、129.29、114.93、112.58、111.68、80.94,70.90、50.84、32.83、29.72、27.32。高分辨质谱数据HRMS(ESI+)for C32H32F3NO5[M+H]+:m/z cale.,567.2233;found,567.2230。
2.GPR40激动活性测试
2.1方法:稳定转染GPR40表达的HEK-293T细胞汇合度达80%后,调整细胞浓度以1×105个/mL的密度接种于96孔培养板,每孔100μL,37℃,5%CO2培养箱内培养24h;弃去培养基,每孔加入HBSS缓冲液轻柔清洗后,加入100μL Fluo-4 AM染料溶液37℃避光孵育30min,染料Fluo-4 AM用含0.1%BSA,20mmol/L HEPES,2.5mmol/L丙磺舒的HBSS配制,终浓度3μmol/L;孵育结束后,用含0.1%BSA,20mmol/L HEPES,2.5mmol/L丙磺舒的HBSS清洗多余染料,并用该HBSS平衡10min;加入不同浓度梯度的化合物,以及激动剂阳性对照GW9508(1μmol/L)、阻断剂阳性对照GW1100(10μmol/L)、阴性对照DMSO(终浓度为0.1%)和空白组(不加任何添加物)。每组3个复孔,用Flexstation仪检测并读取荧光值。每个化合物稀释浓度为:200μmol/L,100μmol/L,50μmol/L,10μmol/L,5μmol/L;
根据所测荧光值按以下公式计算得到每个化合物的EC50值:
相对激动比率=(待测化合物的荧光值-阴性对照的荧光值)/(激动剂阳性对照的荧光值-阴性对照的荧光值)*100%;
抑制率=(阴性对照的荧光值-待测化合物的荧光值)/(阴性对照的荧光值-阻断剂阳性对照的荧光值)*100%。
2.2结果
利用稳定过表达GPR40的HEK-293T细胞对筛选出的候选化合物进行评价,发现其中ADD-16的激动活性最高,其激动活性与GPR40内源性激动剂棕榈酸(Palmitic acid,PA)相当,因此选择其继续进行药理药效学的重点评价。具体候选化合物激动活性见表3。
3.ADD-16对MIN6细胞糖刺激胰岛素分泌的影响(GSIS实验)
3.1方法
MIN6细胞按适宜浓度接种于24孔板中,继续培养至细胞汇合度超过80%后,按以下分组给药:正常对照组(Con)、ADD组(ADD-16给药浓度分别为:100μmol/L、30μmol/L、10μmol/L、3μmol/L、1μmol/L、0.3μmol/L)、TAK875组(给药浓度分别为:10μmol/L、3μmol/L、1μmol/L),每组设置6组复孔。分组给药后继续培养24h,吸出培养基,用无糖KRB缓冲液轻柔冲洗2遍。冲洗后加入无糖KRB缓冲液,37℃孵育10min。孵育结束后,吸出缓冲液,尽量吸尽每孔液体,各组定量加入含16.7mmol/L葡萄糖的KRB缓冲液,37℃孵育1h。1h后吸出孔内所有培养基,2000rmp/min离心20min,收集上清液,-20℃保存。按小鼠胰岛素酶ELISA检测试剂盒说明书操作,检测每组胰岛素含量。
胰岛素酶ELISA检测:取各组培养基上清,按表1步骤操作:
表1操作参数
最终混合物充分振荡混匀后,450nm波长下用酶标仪读取OD值。通过计算得出各组胰岛素含量。
3.2结果
参见图1,从图1中可以看出,ADD-16能够促进MIN6细胞葡萄糖刺激的胰岛素分泌,并且该作用具有浓度依赖性。而100μmol/L的ADD-16因为对MIN6细胞生长产生抑制作用,因而胰岛素分泌较Con组减少。与TAK875对照组相比,发现在相同浓度下,ADD-16的促胰岛素分泌的作用明显优于TAK875。
4.ADD-16对2型糖尿病大鼠糖脂代谢的影响
4.1方法
150只雄性SD大鼠,于洁净恒温(23±2℃)和恒湿(55±10%)动物房中分笼饲养,人工调整每日12h/12h昼夜光循环,自由饮水进食。适应性饲养3天后随机将150只大鼠分为正常组与模型组。正常组(Con,n=15)组大鼠保持原饲料喂养,模型组(n=135)大鼠给予高脂高糖饲料喂养。所有大鼠每日喂食两次,按需调整饲料量,保证饲料充足。分组喂养8周后,模型组大鼠给予腹腔注射25mg/kg的STZ溶液,注射前需禁食12h。注射后继续按原方法喂养,72h尾静脉采血检测血糖,筛选出不同日空腹血糖水平≥11.1mmol/L的大鼠,作为造模成功的人工诱导T2DM大鼠,分组给药进行下一步实验。将造模成功的135只大鼠随机分为9组:(1)T2DM模型组(TD,n=15):每日给予0.5%的CMC-Na溶液灌胃;(2)0.01mg/kg ADD-16给药组(ADD I,n=15):每日给予0.01mg/kg ADD-16溶液灌胃给药;(3)0.1mg/kg ADD-16给药组(ADD II,n=15):每日给予0.1mg/kg ADD-16溶液灌胃给药;(4)1mg/kg ADD-16给药组(ADD III,n=15):每日给予1mg/kg ADD-16溶液灌胃给药;(5)3mg/kg ADD-16给药组(ADDIV,n=15):每日给予3mg/kg ADD-16溶液灌胃给药;(6)10mg/kg ADD-16给药组(ADD V,n=15):每日给予10mg/kg ADD-16溶液灌胃给药;(7)50mg/kg ADD-16给药组(ADD VI,n=15):每日给予50mg/kg ADD-16溶液灌胃给药;(8)二甲双胍对照组(MT,n=15):每日给予250mg/kg二甲双胍溶液灌胃给药;(9)西格列汀对照组(ST,n=15):每日给予6mg/kg西格列汀溶液灌胃给药。同时每日给予Con组0.5%的CMC-Na溶液灌胃,每周监测各组大鼠体重和餐后血糖(postprandial blood sugar,PBG)。实验给药结束后测脂代谢相关指标。
口服葡萄糖耐量实验(OGTT)实验最后一周时,各组大鼠灌胃给药后禁食不禁水16h,检测每只大鼠的空腹血糖值(作为0min)。按每只大鼠体重,给予50%葡萄糖溶液(2g/kg)灌胃给药,给药后开始计时,分别测定15min、30min、60min、90min、120min五个时间点大鼠血糖。120min血糖值测定完成后,恢复饮食。
胰岛素耐量实验(ITT)实验最后一周时,各组大鼠灌胃给药后禁食不禁水5h,检测每只大鼠的空腹血糖值(作为0min),按每只大鼠体重,给予胰岛素(0.8U/kg)皮下注射,分别测定30min、60min、90min、120min四个时间点大鼠血糖。120min血糖值测定完成后,恢复饮食。实验中观察大鼠状态,发生异常时应及时停止实验并给予葡萄糖溶液灌胃,防止出现低血糖引起的死亡
T2DM大鼠血清胰岛素检测大鼠空腹血清胰岛素水平通过商业胰岛素酶联免疫试剂盒检测。机体胰岛素抵抗程度使用胰岛素抵抗指数进行评估,根据文献报道,使用稳态模式评估法计算胰岛素抵抗指数(HOMA-IR),即HOMA-IR=(空腹血糖×空腹胰岛素)/22.5。
4.2结果
4.2.1 ADD-16对人工诱导T2DM大鼠血糖的影响
在ADD III-ADD VI组以及MT、ST组中,大鼠血糖均出现非常明显的下降趋势,其中ST组的血糖降低幅度最为明显。参见图2,从图2可以看出,给药结束后对比各组大鼠血糖值发现,ADD IV、ADD V和ADD VI组大鼠血糖降低幅度最大,与TD组血糖值相比分别降低了55.0%、56.8%和56.2%,比MT组(45.0%)血糖值降低幅度大,与ST组(61.5%)血糖值降低幅度相似,表明ADD-16在3、10、50mg/kg浓度下对T2DM大鼠产生显著降血糖作用。各组大鼠糖化血红蛋白含量变化趋势与餐后血糖值变化趋势基本一致。
4.2.2 ADD-16改善人工诱导T2DM大鼠的葡萄糖耐量异常
从图3可以看出,ADD-16能够提高人工诱导T2DM大鼠的葡萄糖耐受性。
4.2.3 ADD-16改善人工诱导T2DM大鼠胰岛素抵抗
从图4可以看出,ADD-16能够降低T2DM大鼠体内代偿性升高的胰岛素水平,提高大鼠胰岛素敏感性,同时能够在一定程度上促进胰岛素分泌,补足大鼠体内因血糖升高而相对分泌不足的胰岛素水平。ADD-16和阳性对照药都能显著降低HOMA-IR值,即改善T2DM大鼠体内的胰岛素抵抗。
4.2.4 ADD-16改善人工诱导T2DM大鼠的胰岛素耐受性
从图5可以看出,AUC结果显示,与TD组相比ADD IV-ADD VI组以及MT、ST组分别下降了46.5%、43.7%、45.2%、39.9%和51.4%,说明ADD-16和阳性对照药能够改善人工诱导T2DM大鼠的胰岛素耐受性。
4.2.4 ADD-16对人工诱导T2DM大鼠脂代谢紊乱的调节作用
从图6可以看出,与Con组相比,TD组大鼠血清FFA、TC、TG、LDL和HDL水平显著上升(P<0.001),说明高脂高糖饮食加STZ诱导的T2DM大鼠出现了血脂代谢异常。给药干预4周后,ADD III-ADD VI组以及阳性对照药组大鼠血清FFA、TC、TG、LDL和HDL水平明显下降,差异具有统计学意义(P<0.05)。表明ADD-16能够改善人工诱导T2DM大鼠的脂代谢紊乱。有趣的是ADD V(10mg/kg)组大鼠的TG、LDL和HDL水平都明显低于ADD IV(3mg/kg)组大鼠,与两组的降糖作用趋势相反,可以推测出ADD-16在高浓度下具有更好的改善脂代谢紊乱作用,而低浓度具有更优的降糖作用。
5.ADD-16的血浆药代动力学和组织分布学研究
5.1方法
5.1.1血浆动力学研究
健康雄性SD大鼠8只,体重200-220g,置于恒温(23±2℃)和恒湿(55±10%)洁净的动物房中饲养,房间保持12h/12h人工光照循环,自由进食、进水,实验前适应性饲养3天。实验实施前所有大鼠禁食不禁水12h,实验采用单次灌胃给药,给予每只大鼠口服ADD-16溶液(10mg/kg)后,分别于5、10、15、30、45、60、120、240、360、720、1440min时大鼠眼眶处取血,取血后迅速装入加有肝素的离心管内,4℃,3000rpm/min,离心10min,小心吸取上清血浆,-80℃保存。检测时室温自然解冻,按血浆样品预处理步骤操作,采用建立的LC-MS/MS方法测定大鼠单次给药后的血药浓度。
5.1.2组织分布动力学研究
健康雄性SD大鼠40只,体重200-220g,适应性饲养三天,实验前禁食12h。所有大鼠随机分为5组,每组8只。每组大鼠灌胃给予ADD-16溶液(10mg/kg),分别于给药10、30、60、240、480min后麻醉,腹主动脉放血处死。各组大鼠处死后解剖采集心、肝、脾、肺、肾、脑、胰岛等组织,组织样品用生理盐水冲洗干净表面血液后,用滤纸吸干表面水分,称重,-80℃保存。检测时室温自然解冻,采用LC-MS/MS方法测定各组织中ADD-16浓度。
5.2结果
5.2.1血浆动力学结果
从图7可以看出,SD大鼠口服ADD-16溶液后吸收迅速,30min时血药浓度达到最高点。利用DAS 3.0统计软件计算ADD-16的药代动力学参数,其半衰期(t1/2z)约为30.2h,表观分布容积(Vz/F)约为0.36L/kg,清除率(CLz/F)约为0.009L/h/kg,血药浓度最大值(Cmax)约为395.0ng/mL,参见表2。
表2大鼠单次口服ADD-16后药代动力学参数(n=8)
5.2.2参见图8,组织分布结果显示,除肝脏、胰岛外ADD-16在其他组织中的浓度远低于血药浓度,Cmax顺序:肝>胰岛>肺>肾>心>脾>脑,AUC0-8的顺序与Cmax顺序一致。药物在脑组织中浓度最低,表明ADD-16不易透过血脑屏障。药物在肝脏中浓度最高,说明肝脏可能是ADD-16代谢的主要器官。GPR40主要在胰岛β细胞中表达,组织分布试验表明ADD-16具有明确的胰岛靶向性,
6.ADD-16抗T2DM作用的分子机制研究
6.1方法:将MIN6细胞接种于24孔板中,按步骤2.1所述方法培养至细胞汇合度超过80%后,按以下分组给药,每组12个复孔:(1)正常对照组(Con):空白1640培养基培养;(2)胰岛素抵抗模型组(IR):用0.125mmol/L PA处理24h,诱导建立胰岛素抵抗模型;(3)ADD-16给药组(ADD):成功建立胰岛素抵抗模型后,给予10μmol/LADD-16干预24h;(4)TAK875对照组(TAK875):成功建立胰岛素抵抗模型后,给予10μmol/L TAK875作为阳性对照药干预24h;(5)二甲双胍对照组(MT):成功建立胰岛素抵抗模型后,给予10mmol/L二甲双胍作为阳性对照药干预24h;(6)西格列汀对照组(ST):成功建立胰岛素抵抗模型后,给予10μmol/L西格列汀作为阳性对照药干预24h。分组给药结束后吸出培养基,用无糖KRB缓冲液冲洗2遍,再加入无糖KRB缓冲液,37℃孵育30min后,吸出缓冲液,每组一分为二,即每组6个复孔,分别加入含2.8mmol/L或16.7mmol/L葡萄糖的KRB缓冲液,37℃孵育1h后,吸出孔内所有培养基,2000rmp/min离心20min,收集上清液,-20℃保存。检测每组胰岛素含量。
将MIN6细胞接种于24孔板中,按步骤2.1所述方法培养至细胞汇合度超过80%后,按以下分组给药,每组12个复孔:(1)正常对照组(Con):空白1640培养基培养;(2)GW9508组:1μmol/L GW9508干预24h;(3)GW1100组:10μmol/L GW1100干预24h;(4)ADD-16组(ADD):10μmol/LADD-16干预24h;(5)GW1100+GW9508组:10μmol/L GW1100+1μmol/L GW9508干预24h;(6)GW1100+ADD-16组:10μmol/L GW1100+10μmol/LADD-16干预24h。分组给药结束后吸出培养基,用无糖KRB缓冲液冲洗2遍,再加入无糖KRB缓冲液,37℃孵育30min后,吸出缓冲液,每组一分为二,即每组6个复孔,分别加入含2.8mmol/L或16.7mmol/L葡萄糖的KRB缓冲液,37℃孵育1h后,吸出孔内所有培养基,2000rmp/min离心20min,收集上清液,-20℃保存。检测每组胰岛素含量。
将MIN6细胞以适当密度接种于培养皿中,按步骤2.1所述方法培养至汇合度约80%。将细胞分成以下5组:(1)正常对照组(Con):空白1640培养基培养;(2)胰岛素抵抗模型组(IR):用0.125mmol/L PA处理24h,诱导建立MIN6细胞胰岛素抵抗模型;(3)3μmol/LADD-16给药组(ADD I):成功建立胰岛素抵抗模型后,给予3μmol/LADD-16干预24h;(4)10μmol/L ADD-16给药组(ADD II):成功建立胰岛素抵抗模型后,给予10μmol/LADD-16干预24h;(5)TAK875对照组(TAK875):成功建立胰岛素抵抗模型后,给予10μmol/L TAK875作为阳性对照药干预24h。分组给药结束后,收集各组细胞进行WB或IF实验。
6.2结果
6.2.1 ADD-16改善MIN6细胞胰岛素抵抗
与Con组相比IR组细胞的基础胰岛素分泌与高糖刺激胰岛素分泌水平明显降低(P<0.001),而给药组能够明显改善这种抑制现象,除MT组外,ADD、TAK875及ST组胰岛素分泌水平甚至高于Con组(P<0.001),说明ADD-16、TAK875及西格列汀不仅改善了因胰岛素抵抗被抑制的胰岛素分泌,而且刺激MIN6细胞分泌更多胰岛素。在相同给药浓度下,ADD-16的促胰岛素分泌作用略优于TAK875,参见图9中A。对给予GPR40抑制剂GW1100作用的MIN6细胞,ADD-16同样能够增加胰岛素分泌,其作用效果与GPR40激动剂GW9508相似,暗示化合物ADD-16能够通过激活GPR40蛋白发挥促胰岛素分泌作用,参见图9中B。
6.2.2 ADD-16促进胰岛组织中胰岛素信号相关分子表达
PI3K/AKT信号通路是经典的胰岛素信号相关通路,GPR40蛋白激活后能够诱导P38磷酸化,进而促使PGC-1α表达增加,活化的PGC-1α能够促进PPARγ与EP300结合,使EP300磷酸化并进一步激活PPARγ,而PPARγ能够激活PI3K/AKT信号通路,诱导AKT磷酸化,刺激GLUT4向细胞膜易位增加葡萄糖转运摄取。参见图10,通过Western Blot实验,发现与TD组相比,ADD-16能够上调GPR40、PGC-1α、P-P38、P-EP300、PPARγ、P-AKT、PI3K、IRS1以及GLUT4在ZDF大鼠胰岛组织中的表达(P<0.001),且10mg/kg给药组大鼠胰岛组织中胰岛素信号相关分子表达变化最为明显。
6.2.3 ADD-16促进MIN6细胞内胰岛素信号相关分子表达
成功建立MIN6细胞胰岛素抵抗模型后,给予不同浓度ADD-16及TAK875干预24h后,通过Western Blot检测发现ADD-16以及TAK875能够上调GPR40、PGC-1α、P-P38、P-EP300、PPARγ、P-AKT、PI3K、IRS1以及GLUT4在MIN6细胞中的表达(P<0.001),其中10μmol/L ADD-16给药组的蛋白表达变化最为显著(参见图11中A)。通过免疫荧光染色结果也进一步证实ADD-16能够上调GPR40在MIN6细胞中的表达(参见图11中B)。
7.吖啶二酮类化合物与GPR40受体的对接计算
使用ChemBioDraw Ultra14.0绘制32个吖啶二酮类化合物,将其结构式导入Discovery Studio2016中,将化合物的二维结构转化为三维空间结构。使用加氢工具补齐结构式中的氢原子。将这32个化合物分子依据基础结构(ADD-16)进行叠合,赋予MMFF小分子力场,进行能量优化,参数设定为:智能优化200步。
对GPR40受体(PDB id:4PHU)蛋白进行预处理,该晶体复合物结构为GPR40与TAK-875的晶体复合物结构,去除复合物结构中的水分子,补足蛋白质晶体结构中非结构域的缺失氨基酸。
使用基于热区匹配的半柔性分子对接方法(LibDock)进行分子对接,对接运算参数:受体蛋白为进行预处理后的4PHU,配体分子为加氢处理并进行能量智能优化后的32个化合物,对接区域设定为TAK-875所在空间范围(半径13.1808埃米),热区数量设定为100个,对接判定匹配阈值设定为0.25埃米,使用高精密度对接打分默认算法,对接认定判定选best,对接后进行配体分子的能量优化,选取智能优化算法以提高对接的准确性,其余设定保持默认值不变。对接共计得到了1925个对接结果,将每个分子打分最高的结果进行汇总,结果见表3。
表3 32个吖啶二酮化合物与GPR40分子对接结果
参见图12,可以看出表3中的32个化合物均能够与GPR40结合。
综合以上实验可以得出,本发明发现了吖啶二酮类化合物新的药物靶点,即通过激动GPR40受体发挥葡萄糖依赖性的促胰岛素分泌作用,发现了此类合物用于抗糖尿病治疗的先导化合物,开拓了此类化合物在抗2型糖尿病药物中的应用,对于开发抗糖尿病药物具有重要意义。
本发明的化合物ADD-16、ADD-17、ADD-18、ADD-19的活性评价参见表4。
表4 GPR40激动剂的活性评价(μmol/L,n=6)
本发明首次证实了吖啶二酮类化合物激动GPR40受体,参与GPR40-PPARγ-PI3K/Akt-GLUT4信号通路,促进胰岛素分泌,增加肝脏和肌肉组织的葡萄糖消耗,改善胰岛素抵抗,发挥抗2型糖尿病的作用;将吖啶二酮类化合物制备成抗糖尿药物,将为糖尿病的治疗提供全新选择和策略。
本发明为目前2型糖尿病的治疗提供了一种全新的选择和思路,拓宽了抗糖尿病药物的选择领域,也为该技术领域的发展做出了贡献;本发明是具有明确化学结构的化合物,用于制药时可量化投料,有利于制备现代剂型,具有发展成为抗2型糖尿病药物的潜力。
[收起]
附图
评论
全部评论
共{{commentCount}}条{{rs.Msg_Content}}